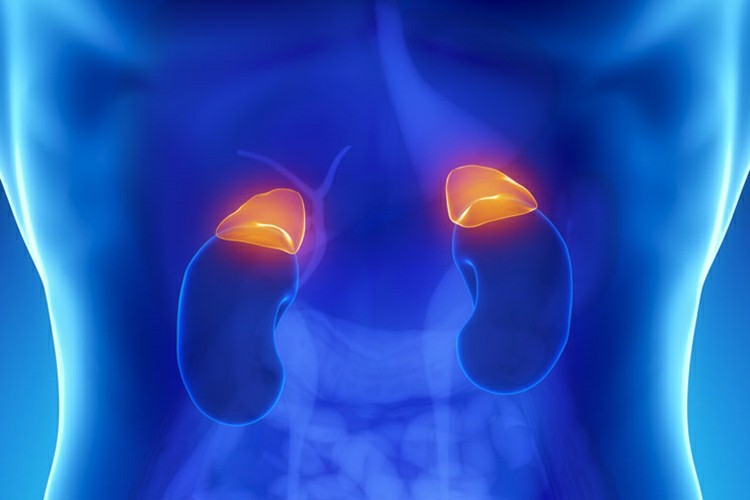
As Principais Causas do Feocromocitoma que não devemos ignorar. Além disso, um Feocromocitoma (PCC) é um tumor raro que pode se formar em células no meio das glândulas supra-renais. No caso do Feocromocitoma, um tumor pode fazer com que as glândulas adrenais façam muito dos hormônios norepinefrina (noradrenalina) e epinefrina (adrenalina). Juntos, esses hormônios controlam a freqüência cardíaca, o metabolismo, a pressão arterial e a resposta ao estímulo do corpo. A maioria do Feocromocitoma cresce dentro das glândulas adrenais. Cerca de 10% crescem nas células de chromafina fora das glândulas supra-renais. Menos de 10% do Feocromocitoma que crescem dentro das glândulas adrenais são cancerígenos, mas esta porcentagem é maior para aqueles que estão fora das glândulas supra-renais.
 Um Feocromocitoma pode ocorrer em homens ou mulheres em qualquer idade, mas são mais comuns em pessoas entre as idades de 20 e 40. Algumas pessoas que desenvolvem Feocromocitoma têm uma condição hereditária rara chamada neoplasia endócrina múltipla que os torna propensos a tumores nas glândulas tireoideia, paratireoide e adrenal.
Um Feocromocitoma pode ocorrer em homens ou mulheres em qualquer idade, mas são mais comuns em pessoas entre as idades de 20 e 40. Algumas pessoas que desenvolvem Feocromocitoma têm uma condição hereditária rara chamada neoplasia endócrina múltipla que os torna propensos a tumores nas glândulas tireoideia, paratireoide e adrenal.
Um Feocromocitoma também pode desenvolver-se em pessoas que têm doença de von Hippel-Lindau e naqueles que apresentam neurofibromatose (doença de von Recklinghausen) ou uma série de outras doenças genéticas. É provável que quase 50% das pessoas que tenham pococromocitomas tenham uma doença genética ou familiar, como estas. Então, confira agora As 6 Principais Causas do Feocromocitoma:
Causas do Feocromocitoma: Os médicos não sabem por que a maioria desses tumores se formam. Cerca de 30%, no entanto, parecem correr em famílias. Estes são mais propensos a ser cancerosos do que aqueles que aparecem aleatoriamente. Eles podem se espalhar para outras partes do seu corpo, incluindo seu fígado , pulmões ou ossos. Os tumores são mais comuns em pessoas com distúrbios ou condições hereditárias, incluindo:
- Neoplasia endócrina múltipla, tipo II
- Doença de Von Hippel-Lindau
- Síndrome do paraganglioma hereditário
- Doença de Von Hippel-Lindau, uma condição em que os cistos e tumores crescem no sistema nervoso central, rins,glândulas adrenais ou outras áreas do corpo
- Neurofibromatose tipo 1, desenvolvimento de tumores na pele e nervos ópticos
- Neoplasia endocrina múltipla tipo 2 (MEN2), uma forma de câncer de tireoide que se desenvolve em conjunto com Feocromocitoma
Sintomas do Feocromocitoma: Um Feocromocitoma pode ser bastante pequenos. No entanto, mesmo um pequeno Feocromocitoma pode produzir grandes quantidades de catecolaminas potentes. As catecolaminas são hormônios como adrenalina ( epinefrina ), norepinefrina e dopamina , que tendem a aumentar significativamente a pressão arterial e a freqüência cardíaca e causar outros sintomas geralmente associados a situações que ameaçam a vida.
O sinal mais proeminente de um Feocromocitoma é a pressão arterial elevada , que pode ser muito grave. No entanto, apenas cerca de 1 em cada 1000 pessoas com hipertensão arterial tem um Feocromocitoma. Os principais sintomas do Feocromocitoma incluem:
VOCÊ JÀ VIU ISSO?
- Dor de cabeça severas
- Suor excessivo
- Respiração rápida
- Pele fria e úmida
- Dor no peito e no estômago
- Náusea e vomito
- Distúrbios da visão
- Dedos formigados
- Prisão de ventre
- Uma estranha sensação de destruição iminente
Quando esses sintomas aparecem de forma repentina e com força, eles podem se sentir como um ataque de pânico . Na metade das pessoas afetadas, os sintomas irão e vir, às vezes desencadeados pela pressão sobre o tumor, massagem, drogas (especialmente anestesias e beta-bloqueadores), trauma emocional e, em raras ocasiões, o simples ato de urinar. No entanto, muitas pessoas podem ter esses sintomas como manifestações de um estado de ansiedade, e não uma doença glandular.
Diagnóstico do Feocromocitoma: Muitas pessoas que têm Feocromocitoma nunca são diagnosticadas porque os sintomas são muito semelhantes aos de outras condições. Os principais diagnóstico do Feocromocitoma incluem:
- Testes de sangue e urina
- Tomografia computadorizada ou ressonância magnética
Os médicos podem não suspeitar de um Feocromocitoma, porque quase metade das pessoas não apresentam sintomas além da hipertensão arterial persistente. No entanto, quando a pressão arterial elevada ocorre em uma pessoa jovem, vai e vem, ou acompanha outros sintomas de Feocromocitoma, os médicos podem solicitar determinados testes laboratoriais.
Por exemplo, o nível de determinadas catecolaminas ou produtos criados quando estas catecolaminas são discriminadas pode ser medido em amostras de sangue ou urina. Devido à pressão arterial elevada e outros sintomas, os médicos podem prescrever um betabloqueador antes de saber que a causa é um Feocromocitoma. Os beta-bloqueadores podem piorar a pressão arterial em pessoas com Feocromocitoma. Essa reação paradoxal muitas vezes torna claro o diagnóstico de Feocromocitoma.
Se o nível de catecolaminas for alto, a tomografia computadorizada (TC) ou a ressonância magnética (MRI) podem ajudar a localizar o Feocromocitoma. Um teste com produtos químicos radioativos injetados que tendem a se acumular em Feocromocitoma também pode ser útil. Uma varredura é então feita para ver onde os produtos químicos radioativos são. Testes genéticos podem ser feitos, especialmente se os médicos suspeitarem de uma doença genética.
Tratamentos do Feocromocitoma: Os principais tratamentos do Feocromocitoma incluem:
- Cirurgia para remover o tumor
- Medicamentos para controlar a pressão sanguínea.
Normalmente, o melhor tratamento é remover o Feocromocitoma. A cirurgia muitas vezes está atrasada, no entanto, até que os médicos possam controlar a secreção de catecolaminas do tumor com drogas, pois ter níveis elevados de catecolaminas pode ser perigoso durante a cirurgia. A fenoxibenzamina ou um fármaco similar é geralmente administrado para parar a ação hormonal.
Uma vez que este passo é realizado, um bloqueador beta pode ser administrado com segurança a outros sintomas de controle. Se o Feocromocitoma é cancerígeno e se espalhou, a quimioterapia raramente cura o tumor. Novas drogas de quimioterapia, como temozolomida e sunitinib, parecem ajudar a diminuir o crescimento do tumor.
O tratamento com um radioisótopo conhecido como metaiodobenzilguanidina (MIBG) que visa o tecido tumoral também pode ser altamente eficaz. Os efeitos perigosos das catecolaminas em excesso segregadas pelo tumor podem quase sempre ser bloqueados continuando a tomar fenoxibenzamina ou um fármaco e betabloqueadores semelhantes.