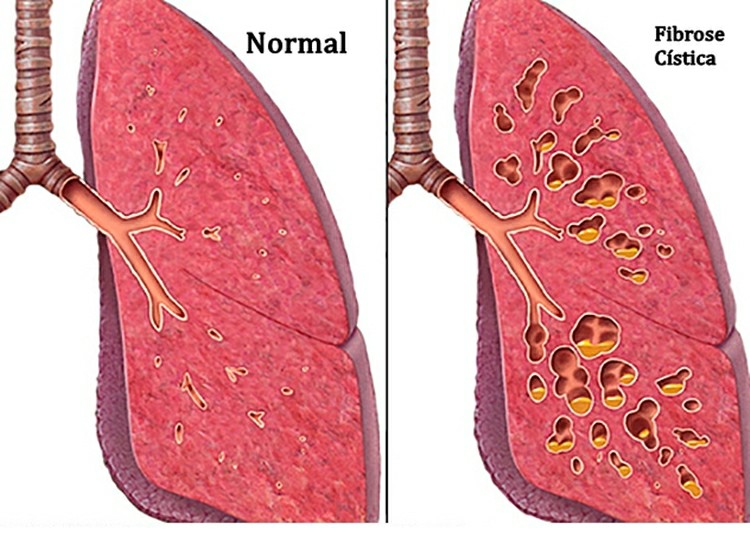
Fibrose Cística – O que é, Sintomas e Tratamentos com antibióticos. Além disso, a Fibrose Cística é uma desordem hereditária que causa danos graves aos pulmões, sistema digestivo e outros órgãos no corpo. A Fibrose Cística afeta as células que produzem muco, suor e sucos digestivos. Estes fluidos secretos são normalmente finos e escorregadios. Mas em pessoas com Fibrose Cística, um gene defeituoso faz com que as secreções se tornem pegajosas e grossas. Em vez de atuar como um lubrificante, as secreções conectam tubos, dutos e passagens, especialmente nos pulmões e no pâncreas.
Embora a Fibrose Cística requer cuidados diários, as pessoas com a condição geralmente são capazes de frequentar a escola e do trabalho, e muitas vezes têm uma melhor qualidade de vida do que as pessoas com Fibrose Cística nas décadas anteriores. Melhorias na triagem e nos tratamentos significam que pessoas com Fibrose Cística agora podem viver em meados dos anos 30, em média, e algumas estão vivendo em seus 40 e 50 anos.
Causas de Fibrose Cística: Na Fibrose Cística, um defeito (mutação) em um gene muda uma proteína que regula o movimento do sal dentro e fora das células. O resultado é mucosa grossa e pegajosa nos sistemas respiratório, digestivo e reprodutivo, bem como aumento do sal no suor.
Muitos defeitos diferentes podem ocorrer no gene. O tipo de mutação genética está associada à gravidade da condição. As crianças precisam herdar uma cópia do gene de cada pai para ter a doença. Se os filhos herdam apenas uma cópia, eles não desenvolverão Fibrose Cística. No entanto, eles serão transportadores e possivelmente passarão o gene para seus próprios filhos.
Sintomas de Fibrose Cística: O rastreio de recém-nascidos para Fibrose Cística é agora realizado em todos os estados nos Estados Unidos. Como resultado, a condição pode ser diagnosticada no primeiro mês de vida, antes que os sintomas se desenvolvam. Para as pessoas nascidas antes do rastreio do recém-nascido, é importante estar ciente dos sinais e sintomas da Fibrose Cística. Os sinais e sintomas de Fibrose Cística variam, dependendo da gravidade da doença. Mesmo na mesma pessoa, os sintomas podem piorar ou melhorar à medida que o tempo passa. Algumas pessoas podem não sentir sintomas até a adolescência ou a idade adulta.
As pessoas com Fibrose Cística têm um nível de sal elevado ao normal em seu suor. Os pais geralmente podem saborear o sal quando beijam seus filhos. A maioria dos outros sinais e sintomas de Fibrose Cística afetam o sistema respiratório eo sistema digestivo. No entanto, adultos com diagnóstico de Fibrose Cística são mais propensos a ter sintomas atípicos, tais como episódios recorrentes de pâncreas inflamadas (pancreatite), infertilidade e pneumonia recorrente.
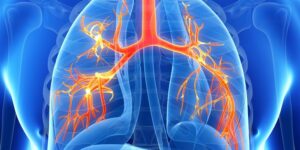
Sinais e Sintomas Respiratórios: O muco grosso e pegajoso associado à Fibrose Cística obstrui os tubos que transportam ar dentro e fora de seus pulmões. Isso pode causar sinais e sintomas como:
- Uma tosse persistente que produz muco espesso (escarro)
- Sibilância
- Falta de ar
- Intolerância ao exercício
- Infecções pulmonares repetidas
- Passagens nasais inflamadas ou nariz entupido
Sinais e Sintomas Digestivos: O muco grosso também pode bloquear os tubos que transportam enzimas digestivas do seu pâncreas para o intestino delgado. Sem essas enzimas digestivas, seus intestinos não conseguem absorver completamente os nutrientes nos alimentos que você come. O resultado é freqüentemente:
- Fezes com cheiro mal cheiro e fezes gordurosas
- Maior aumento de peso e crescimento
- Bloqueio intestinal, particularmente em recém-nascidos (íleo do mecônio)
- Constipação grave
O esforço freqüente ao passar fezes pode causar parte do reto – o fim do intestino grosso – para protrusão fora do ânus (prolapso retal). Quando isso ocorre em crianças, pode ser um sinal de Fibrose Cística. Os pais devem consultar um médico experiente em Fibrose Cística. O prolapso retal em crianças às vezes pode requerer cirurgia. O prolapso retal em crianças com Fibrose Cística é menos comum do que no passado, o que pode ser devido a testes anteriores, diagnóstico e tratamento da Fibrose Cística.
Quando Consultar um Médico: Se você ou o seu filho tiverem sintomas de Fibrose Cística – ou se alguém na sua família tiver Fibrose Cística – fale com o seu médico sobre o teste da doença. Procure cuidados médicos imediatos se você ou seu filho tiverem dificuldade em respirar.
Fatores de Risco de Fibrose Cística: História de família. Como a Fibrose Cística é uma desordem hereditária, ela é executada em famílias.
- Corrida. Embora a Fibrose Cística ocorra em todas as raças, é mais comum em pessoas brancas de ascendência do norte da Europa.
Complicações de Fibrose Cística: Complicações do sistema respiratório
- Vias aéreas danificadas (bronquiectasias): A Fibrose Cística é uma das principais causas de bronquiectasias, uma condição que danifica as vias aéreas. Isso torna mais difícil mover o ar dentro e fora dos pulmões e limpar o muco das vias aéreas (tubos brônquicos).
- Infecções Crônicas: O muco grosseiro nos pulmões e nos seios fornece um terreno propágulo ideal para bactérias e fungos. Pessoas com Fibrose Cística geralmente podem ter infecções sinusais, bronquite ou pneumonia.
- Crescimentos no nariz (pólipos nasais): Como o revestimento dentro do nariz está inflamado e inchado, ele pode desenvolver crescimentos suaves e carnudos (pólipos).
- Tossindo sangue (hemoptise): Ao longo do tempo, a Fibrose Cística pode causar desbaste das paredes das vias aéreas. Como resultado, adolescentes e adultos com Fibrose Cística podem tossir sangue.
- Pneumotórax: Esta condição, na qual o ar se acumula no espaço que separa os pulmõess da parede torácica, também é mais comum em pessoas idosas com Fibrose Cística. Pneumotórax pode causar dor torácica e falta de ar.
- Parada Respiratória: Ao longo do tempo, a Fibrose Cística pode danificar tanto o tecido pulmonar que não funciona mais. A função pulmonar geralmente piora gradualmente, e eventualmente pode tornar-se fatal.
- Exacerbações Agudas: Pessoas com Fibrose Cística podem sofrer agravamento de seus sintomas respiratórios, como tosse e falta de ar, durante vários dias a semanas. Isso é chamado de exacerbação aguda e requer tratamento no hospital.
Complicações do sistema digestivo:
- Deficiências Nutricionais: O muco grosso pode bloquear os tubos que transportam enzimas digestivas do seu pâncreas para os intestinos. Sem essas enzimas, seu corpo não pode absorver proteínas, gorduras ou vitaminas solúveis em gordura.
- Diabetes: O pâncreas produz insulina, que seu corpo precisa usar o açúcar. A Fibrose Cística aumenta o risco de diabetes. Cerca de 30 por cento das pessoas com Fibrose Cística desenvolvem diabetes até os 30 anos.
- Duto Biliar Bloqueado: O tubo que transporta a bile do seu fígado e vesícula até o intestino delgado pode ficar bloqueado e inflamado, levando a problemas hepáticos e às vezes cálculos biliares.
- Obstrução Intestinal: Obstrução intestinal pode acontecer a pessoas com Fibrose Cística em todas as idades. Crianças e adultos com Fibrose Cística são mais propensos do que os bebês a desenvolver intussuscepção, uma condição em que uma seção dos intestinos se dobra em si mesma como um acordeão.
- Síndrome de Obstrução Intestinal Distal (DIOS): DIOS é obstrução parcial ou completa onde o intestino delgado encontra o intestino grosso.
Complicações do sistema reprodutivo: Quase todos os homens com Fibrose Cística são inférteis porque o tubo que liga os testículos e próstata (vasos deferentes) é bloqueado com muco ou falta completamente. Certos tratamentos de fertilidade e procedimentos cirúrgicos às vezes tornam possível que homens com Fibrose Cística se tornem pais biológicos.
VOCÊ JÀ VIU ISSO?
Embora as mulheres com Fibrose Cística possam ser menos férteis do que outras mulheres, é possível para elas conceber e ter gravidezes bem-sucedidas. Ainda assim, a gravidez pode piorar os sinais e sintomas da Fibrose Cística, por isso não deixe de discutir os possíveis riscos com o seu médico. Outras complicações:
- Diluição dos Ossos (Osteoporose): As pessoas com Fibrose Cística correm maior risco de desenvolver um desbaste perigoso de ossos.
- Desequilíbrios Eletrolíticos e Desidratação: Como as pessoas com Fibrose Cística têm suor mais salgado, o equilíbrio de minerais no sangue pode estar chateado. Sinais e sintomas incluem aumento da freqüência cardíaca, fadiga, fraqueza e baixa pressão arterial.
Diagnóstico de Fibrose Cística: Para diagnosticar Fibrose Cística, os médicos podem realizar vários testes. Rastreio e diagnóstico do recém-nascido: Todos os estados nos EUA agora rotulam os recém-nascidos para Fibrose Cística. O diagnóstico precoce significa que o tratamento pode começar imediatamente.

Em um teste de triagem, uma amostra de sangue é verificada por níveis mais altos do que o normal de um produto químico (tripsinogênio imunorreativo, ou IRT) liberado pelo pâncreas. Os níveis de IRT de um recém-nascido podem ser elevados devido ao nascimento prematuro ou a uma entrega estressante. Por esse motivo, outros testes podem ser necessários para confirmar o diagnóstico de Fibrose Cística.
Testes genéticos podem ser usados além de verificar os níveis de IRT para confirmar o diagnóstico. Os médicos também podem realizar testes genéticos para testar defeitos específicos no gene responsável pela fibrose cística. Para avaliar se uma criança tem fibrose cística, os médicos também podem realizar um teste de suor quando a criança tiver pelo menos 2 semanas de idade.
Em um teste de suor, os médicos aplicam um produto químico produtor de suor a uma pequena área da pele. Eles então coletam o suor para testá-lo e ver se ele é mais salgado do que o normal. Os testes podem ser feitos em um centro especializado em Fibrose Cística.
Testes de Crianças e Adultos mais Velhos: Testes de Fibrose Cística podem ser recomendados para crianças mais velhas e adultos que não foram selecionados no nascimento. Seu médico pode sugerir testes genéticos e de suor para fibrose cística se você tiver episódios recorrentes de pâncreas inflamadas (pancreatite), pólipos nasais, sinusite crônica ou infecções pulmonares, bronquiectasias ou infertilidade masculina.
Tratamentos de Fibrose Cística: Não há cura para a Fibrose Cística, mas o tratamento pode aliviar os sintomas e reduzir as complicações. É recomendado um monitoramento próximo e uma intervenção agressiva precoce. O gerenciamento da Fibrose Cística é complexo, portanto, considere a obtenção de tratamento em um centro formado por médicos e outras equipes treinadas em Fibrose Cística.
Os médicos podem trabalhar com uma equipe multidisciplinar de médicos e profissionais médicos treinados em Fibrose Cística para avaliar e tratar sua condição. Os objetivos do tratamento incluem:
- Prevenção e controle de infecções que ocorrem nos pulmões
- Removendo e afrouxando o muco dos pulmões
- Tratamento e prevenção do bloqueio intestinal
- Fornecer nutrição adequada
Medicamentos: As opções podem incluir:
- Antibióticos para tratar e prevenir infecções pulmonares
- Medicamentos anti-inflamatórios para diminuir o inchaço nas vias aéreas nos pulmões
- Medicamentos de amassar o muco para ajudá-lo a tossir o muco, o que pode melhorar a função pulmonar
- Medicamentos inalados chamados de broncodilatadores que podem ajudar a manter as vias respiratórias abertas, .relaxando os músculos ao redor de seus brônquios
- Enzimas pancreáticas orais para ajudar seu aparelho digestivo a absorver nutrientes
Para aqueles com Fibrose Cística que possuem certas mutações genéticas, os médicos podem recomendar um medicamento mais recente chamado ivacaftor (Kalydeco). Este medicamento pode melhorar a função e o peso pulmonares e reduzir a quantidade de sal no suor. Foi aprovado pela Food and Drug Administration para pessoas com Fibrose Cística com idade igual ou superior a 6 anos. A dose depende do seu peso e idade.
Os médicos podem realizar exames de função hepática e exames oculares antes de prescrever-se e, regularmente, enquanto você o está tomando para verificar se há efeitos colaterais como anormalidades da função hepática e cataratas. Para pessoas com uma certa mutação genética com idade entre 12 e mais anos, outra droga (Orkambi) está disponível que combina ivacaftor com uma medicação chamada lumacaftor.
A combinação destes medicamentos pode melhorar a função pulmonar e reduzir o risco de exacerbações. No entanto, algumas pessoas podem experimentar efeitos colaterais, como desconforto no tórax e falta de ar, logo após o início da medicação. Algumas pessoas também podem ter alta pressão arterial enquanto tomam a medicação. Os médicos podem acompanhá-lo por quaisquer efeitos colaterais.
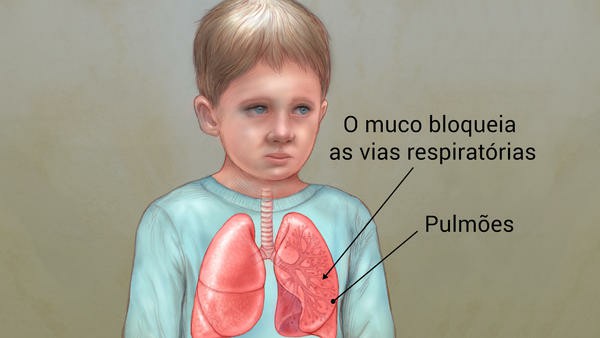 Fisioterapia no Tórax: Afrouxar o muco grosso nos pulmões facilita a tosse. A terapia física do tórax ajuda a afrouxar o muco. Geralmente é feito de uma a quatro vezes por dia. Uma técnica comum é aplaudir com as mãos colocadas na frente e na parte de trás do baú. Certas técnicas de respiração também podem ser usadas para ajudar a afrouxar o muco. O seu médico irá instruí-lo sobre o tipo de fisioterapia no tórax que ele ou ela recomenda para você.
Fisioterapia no Tórax: Afrouxar o muco grosso nos pulmões facilita a tosse. A terapia física do tórax ajuda a afrouxar o muco. Geralmente é feito de uma a quatro vezes por dia. Uma técnica comum é aplaudir com as mãos colocadas na frente e na parte de trás do baú. Certas técnicas de respiração também podem ser usadas para ajudar a afrouxar o muco. O seu médico irá instruí-lo sobre o tipo de fisioterapia no tórax que ele ou ela recomenda para você.
Reabilitação Pulmonar: O seu médico pode recomendar um programa a longo prazo que possa melhorar a sua função pulmonar e bem-estar geral. A reabilitação pulmonar geralmente é feita de forma ambulatorial e pode incluir:
- Exercício físico que pode melhorar sua condição
- Técnicas de respiração que podem ajudar a afrouxar o muco e melhorar a respiração
- Aconselhamento nutricional
- Aconselhamento e apoio
- Educação sobre sua condição
Procedimentos cirúrgicos e outros:
- Remoção de pólipos nasais. Seu médico pode recomendar cirurgia para remover pólipos nasais que obstruem a respiração.
- Terapia de oxigênio. Se o seu nível de oxigênio no sangue diminui, seu médico pode recomendar que você respire oxigênio puro para prevenir a pressão arterial elevada nos pulmões (hipertensão pulmonar).
- Endoscopia e lavagem. Mucus pode ser aspirado de vias aéreas obstruídas através de um endoscópio.
- Tubo de alimentação. A Fibrose Cística interfere na digestão, de modo que você não consegue absorver alimentos muito bons. Seu médico pode sugerir temporariamente usar um tubo de alimentação para fornecer nutrição extra enquanto você dorme. Este tubo pode ser inserido no nariz e guiado para o estômago, ou pode ser implantado cirurgicamente no abdômen.
- Cirurgia intestinal. Se um bloqueio se desenvolver em seu intestino, você pode precisar de cirurgia para removê-lo. Intussuscepção, onde uma seção de intestino dobrou sobre si mesma, também pode exigir reparo cirúrgico.
- Transplante pulmonar. Se você tiver problemas respiratórios graves, complicações pulmonares com risco de vida ou aumento da resistência aos antibióticos utilizados para tratar infecções pulmonares, o transplante pulmonar pode ser uma opção. Como as bactérias alinham as vias aéreas em doenças que causam o alargamento permanente das grandes vias aéreas (bronquiectasias), como a Fibrose Cística, ambos os pulmões precisam ser substituídos.
A Fibrose Cística não se repete nos pulmões transplantados. No entanto, outras complicações associadas à Fibrose Cística – como infecções dos sinusites, diabetes, problemas do pâncreas e osteoporose – ainda podem ocorrer após um transplante de pulmão.